In general, solids are characterized by structural rigidity and resistance to changes in shape or volume. Unlike a liquid, a solid object does not flow to take on the shape of its container, nor does it expand to fill the entire volume available to it like a gas does. Solids have greater interatomic attractions than liquids and gases. However, there are wide variations in the properties of solid materials used for engineering purposes. The properties of materials depend on their interatomic bonds, and these same bonds also dictate the space between the configuration of atoms in solids. All solids may be classified as:
- Amorphous solids. Amorphous materials, or non-crystalline solids, have no regular arrangement of their molecules and lack the long-range order characteristic of crystalline solids. Amorphous materials have the properties of solids and are characterized by structural rigidity and resistance to changes in shape or volume. They have definite shape and volume and diffuse slowly. Materials like glass and paraffin are considered amorphous, and these materials also lack sharply defined melting points. In many respects, they resemble liquids that flow very slowly at room temperature.
- Crystalline solids. The atoms in a crystalline solid are tightly bound to each other in a regular geometric lattice (crystalline solids, which include metals and ordinary ice).

A crystalline material is one in which the atoms are situated in a repeating or periodic array over large atomic distances. That is, long-range order exists. Upon solidification, the atoms will position themselves in a repetitive three-dimensional pattern, in which each atom is bonded to its nearest neighbor atoms. Not all solids are single crystals. For example, when liquid water starts freezing, the phase change begins with small ice crystals that grow until they fuse, forming a polycrystalline structure. In the final block of ice, each of the small crystals (called “grains“) is a true crystal with a periodic arrangement of atoms. Still, the whole polycrystal does not have a periodic arrangement of atoms because the periodic pattern is broken at the grain boundaries.
Crystal Lattice
Some of the properties of crystalline solids depend on the crystal structure of the material and how atoms, ions, or molecules are spatially arranged. A crystal lattice is a repeating pattern of mathematical points that extends throughout space, and the forces of chemical bonding cause this repetition. This repeated pattern controls properties like strength, ductility, density, conductivity (property of conducting or transmitting heat, electricity, etc.), and shape. There are 14 general types of such patterns known as Bravais lattices.
Three relatively simple crystal structures are found for most of the common metals:
-
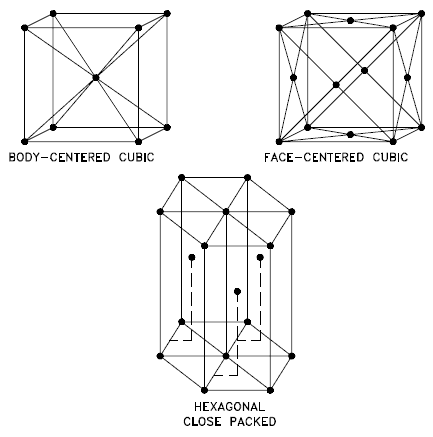
Source: U.S. Department of Energy, Material Science. DOE Fundamentals Handbook, Volume 1 and 2. January 1993. Body-centered Cubic – bcc. In a bcc (BCC) arrangement of atoms, the unit cell consists of eight atoms at the corners of a cube and one atom at the body center of the cube. In a bcc arrangement, a unit cell contains (8 corner atoms × ⅛) + (1 center atom × 1) = 2 atoms. The packing is more efficient (68%) than simple cubic, and the structure is common for alkali and early transition metals. Metals containing BCC structures include ferrite, chromium, vanadium, molybdenum, and tungsten. These metals possess high strength and low ductility.
- Face-centered Cubic – fcc. In an fcc (FCC) arrangement of atoms, the unit cell consists of eight atoms at the corners of a cube and one atom at the center of each of the faces of the cube. In an fcc arrangement, a unit cell contains (8 corner atoms × ⅛) + (6 face atoms × ½) = 4 atoms. This structure, along with its hexagonal relative (hcp), has the most efficient packing (74%). Metals containing FCC structures include austenite, aluminum, copper, lead, silver, gold, nickel, platinum, and thorium. These metals possess low strength and high ductility.
- Hexagonal Close-packed – hcp. In an hcp (HCP) arrangement of atoms, the unit cell consists of three layers of atoms. The top and bottom layers contain six atoms at the corners of a hexagon and one atom at the center of each hexagon. The middle layer contains three atoms nestled between the atoms of the top and bottom layers. Hence, the name is close-packed. Hexagonal close-packed (hcp) is one of the two simple types of atomic packing with the highest density, the other being the face-centered cubic (fcc). However, unlike the fcc, it is not a Bravais lattice as there are two nonequivalent sets of lattice points. Metals containing HCP structures include beryllium, magnesium, zinc, cadmium, cobalt, thallium, and zirconium. HCP metals are not as ductile as FCC metals.

Atomic Packing Factor – APF
In crystallography, atomic packing factor (APF), packing efficiency, or packing fraction is the sum of the sphere volumes of all atoms within a unit cell (assuming the atomic hard-sphere model) divided by the unit cell volume.
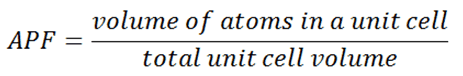
By convention, the APF is determined by assuming that atoms are rigid spheres, and the radius of the spheres is taken to be the maximum value such that the atoms do not overlap. In materials science, the atomic packing factor of a unit cell explains many properties of materials. For example, metals with a high atomic packing factor will have a higher malleability or ductility, similar to how a road is smoother when the stones are closer together, allowing metal atoms to slide past one another more easily.
For example, in an fcc arrangement, a unit cell contains (8 corner atoms × ⅛) + (6 face atoms × ½) = 4 atoms. This structure, along with its hexagonal relative (hcp), has the most efficient packing (74%).
Grain Structure and Grain Boundaries

Not all solids are single crystals (e.g., silicon semiconductors). Most crystalline solids are composed of a collection of many small crystals or grains of varying sizes and orientations, and these have random crystallographic orientations. When metal starts with crystallization, the phase change begins with small crystals that grow until they fuse, forming a polycrystalline structure. In the final block of solid material, each of the small crystals (called “grains“) is a true crystal with a periodic arrangement of atoms. Still, the whole polycrystal does not have a periodic arrangement of atoms because the periodic pattern is broken at the grain boundaries. Grains and grain boundaries help determine the properties of a material.
- Grains, also known as crystallites, are small or microscopic crystals that form, for example, during the cooling of many materials (crystallization). A very important feature of a metal is the average size of the grain, and the size of the grain determines the properties of the metal. For example, smaller grain size increases tensile strength and tends to increase ductility. Larger grain size is preferred for improved high-temperature creep properties. Creep is the permanent deformation that increases with time under constant load or stress, and creep becomes progressively easier with increasing temperature.
- Grain Boundaries. The grain boundary refers to the outside area of a grain that separates it from the other grains. The grain boundaries separate variously-oriented crystal regions (polycrystalline) in which the crystal structures are identical. Grain boundaries are 2D defects in the crystal structure and tend to decrease the electrical and thermal conductivity of the material. Most grain boundaries are preferred sites for the onset of corrosion and the precipitation of new phases from the solid. They are also important to many of the mechanisms of creep. On the other hand, grain boundaries disrupt the motion of dislocations through a material. Dislocation propagation is impeded because of the grain boundary defect region’s stress field, the lack of slip planes and slip directions, and overall alignment across the boundaries. Therefore reducing crystallite size is a common way to improve mechanical strength because the smaller grains create more obstacles per unit area of the slip plane.
Grains Orientation
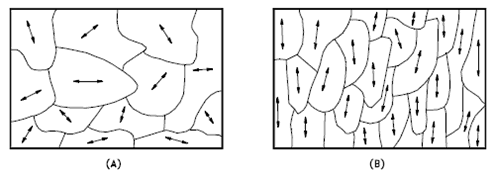
The orientation of crystallites can be random with no preferred direction, called random texture, or preferred, possibly due to growth and processing conditions. Random orientation can be obtained by cross-rolling the material. If such a sample were rolled sufficiently in one direction, it might develop a grain-oriented structure in the rolling direction, which is called preferred orientation. In many cases, preferred orientation is desirable, but it can be most harmful in other instances. For example, preferred orientation in uranium fuel elements can result in catastrophic changes in dimensions during use in a nuclear reactor.
Special reference: U.S. Department of Energy, Material Science. DOE Fundamentals Handbook, Volume 1 and 2. January 1993.
Polymorphism and Allotropy
Some crystalline materials may have more than one crystal structure, a phenomenon known as polymorphism. Polymorphism is the occurrence of multiple crystalline forms of a material. According to Gibbs’ rules of phase equilibria, these unique crystalline phases depend on intensive variables such as pressure and temperature. Polymorphism is related to allotropy, which refers to chemical elements. That means polymorphism is the more general term for any crystalline material, including alloys and chemical elements. Each polymorph is a different thermodynamic solid state, and crystal polymorphs of the same compound exhibit different physical properties, such as dissolution rate, shape (angles between facets and facet growth rates), melting point, etc. For this reason, polymorphism is of major importance in the industrial manufacture of crystalline products.
One familiar example is found in carbon. Graphite (a soft, black, flaky solid, a moderate electrical conductor) is the stable polymorph under ambient conditions, whereas diamond (an extremely hard, transparent crystal with carbon atoms arranged in a tetrahedral lattice. A poor electrical conductor. An excellent thermal conductor.) is formed at extremely high pressures.
Also, pure iron has a BCC crystal structure at room temperature, which changes to FCC iron at 913 °C. Zirconium is HCP (alpha) up to 863°C, where it transforms to the BCC (beta, zirconium) form. The problem is that these phase transitions make this material very brittle.
See also: High-Temperature Steam Oxidation of Zirconium Alloys